



Cuantificación relativa normalizada respecto un gen de referenciaHabitualmente, la cuantificación relativa se utiliza para conocer si la expresión de un gen específico varia entre dos grupos, por ejemplo: grupo control vs grupo tratado.Para obtener una cuantificación relativa que se ajuste al máximo a la realidad, los datos obtenidos de la Real-time RT-PCR deben normalizarse. Seguir unas estartegias de normalización apropiadas permitirá controlar posibles errores introducidos a lo largo del proceso experimental. En la cuantificación relativa podemos diferenciar entre dos métodos de normalización:
Gen de referenciaEl control interno o gen de referencia se caracteriza por presentar una expresión constante y invariable en todas las muestras. La expresión de este no puede ser modificada por las condiciones de estudio o por la respuesta a un tratamiento experimental. La literatura nos muestra que no se han identificado genes de referencia óptimos (los genes utilizados habitualmente varian considerablemente entre muestras), sin embargo este método de normalización es el mas utilizado. En consequencia, la herramienta desenvolupada implementa un método específico para identificar los genes control óptimos, conocido como Vandesompele.El método Vandesompele se basa en la estabilidad de los genes para identificar el gen de referencia, como mas estable sea un gen mejor gen de referencia será. De modo que se obtiene un ranking con los genes mas estables, permitiendo el uso de mas de un gen de referencia si se da el caso (recomendable utilizar 2 genes de referencia para la normalización). Una vez identificados los genes con mejor estabilidad, se realiza la media geométrica de estos a partir de la qual se normalizaran los datos. Cuantifiación relativaSeleccionado el gen de referencia se prosigue con la cuantificación relativa.Para calcular la expresión relativa a un gen de referencia disponemos de tres métodos diferentes: 2-ΔΔCT Method, 2-ΔCT Method y Pfaffl Method. La herramienta CT utiliza una fórmula matemática derivada de Pfaffl Method para el cálculo de la expresión relativa (esta es similar al método presentado por Soong et al. -Roche Diagnostics (2000)-). El algoritmo matemático derivado de Pfaffl method utilizado se define como: 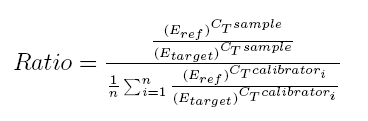 Respecto otros métodos este permite el cálculo de la expresión relativa teniendo en cuenta la eficiencia de cada gen y utilizando mas de una muestra calibradora. Eficiencia de la Real-time RT-PCRDe forma general, la PCR consiste en cuatro etapas, similares a las de la cinética de crecimiento bacteiano observada por Monod. En la segunda etapa de esta se da una fase logarítmica, equivalmente al crecimiento exponencial del producto de la PCR --idelamente D.O. del producto de PCR se dobla en cada ciclo-- mesurado como señal de fluorescencia. La cinética de la PCR en esta fase puede descrivirse como:Así, el cálculo de la eficiencia por un gen específico se obtinen de: Eff=10slope. En la literatura se ha descrito como el uso por defecto de valores de eficiencia de 2 (100% eficiencia de la PCR) afecta significativamente al cálculo acurado de los niveles de expresión relativos a un gen específico; observando hasta un error de 4-fold cuando la eficiencia varia tan solo en un rango de 0,04. Para calcular la eficiencia de cada gen, la herramienta CT utiliza los datos de fluorescencia mesurados por cada ciclo a lo largo de todo el proceso de amplificación. El algoritmo implementado por el cálculo de la eficiencia se basa en el programa LinRegPCR. Concretamente, el proceso para obtener la eficiencia consiste en linearizar los datos de la fase exponencial de la PCR y determinar la mejor regresión lineal. Este proceso se realiza por cada gen y cada muestra, obteniendo un resultado específico por cada caso. Los valores finales de eficiencia por gen son el resultado de la media de los diferentes valores de cada gen. 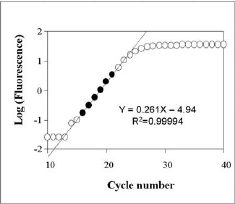
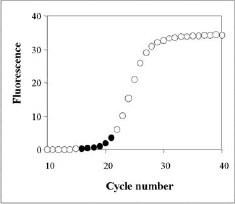
En las dos figuras que se muestran a la derecha representan el funcionamiento del programa para obtener el valor de eficiencia. |
||

